Am citit un articol care trimitea catre un alt articol de tabloid din UK.
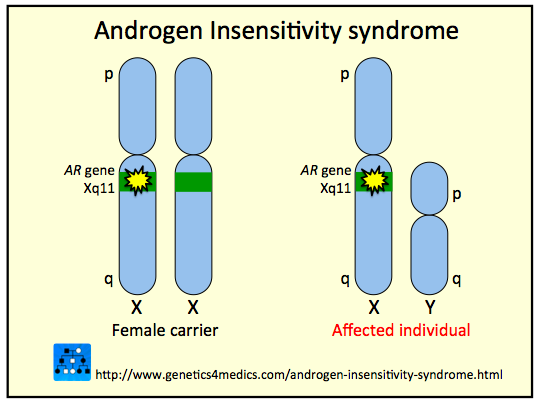
Povestea, pe scurt, ar fi: o femeie este diagnosticata la 19 ani cu un sindrom rar, numit Sindrom de Insensibilitate la hormonii Androgeni (AIS), mai precis, cu forma “completa” a AIS. Asta inseamna ca, desi aspectul sau era de femeie, este, din punct de vedere genetic, un barbat. Un barbat asupra caruia hormonii androgeni nu si-au facut efectul si nu i-au dezvoltat trasaturile masculine. Adica cromozomii ei 45 si 46 erau de fapt XY (barbat), nu XX (femeie). Cu toate ca i s-a spus ca nu va putea concepe si naste niciodata copii, la 9 ani de la diagnostic ea tocmai a dat nastere unui cuplu de gemeni. Cum a fost posibil?
AIS se manifesta prin dezvoltarea caracteristicilor feminine ale copilului mascul inca din burta mamei, majoritar din cauza unei malformatii a genei AR (care codeaza in anumite celule in mod normal proteina Androgen Receptor, responsabila pentru declansarea procesului de masculinizare) sau din cauza altor mutatii ale unor gene care codeaza proteine importante pentru recunoasterea hormonilor androgeni de catre tesuturi.
Rezultatul (mai ales in forma completa a AIS) este un adult care arata 100% ca o femeie, dar are vaginul de multe ori incomplet, uterul, ovarele si trompele uterine lipsesc cu desavarsire sau sunt minuscule, penisul lipseste iar testiculele lipsesc sau nu coboara niciodata in scrot (care poate lipsi, la randul sau). Este evident ca o persoana cu tipul complet de sindrom este infertila (nu are celule reproducatoare). Exista si o varianta blanda, respectiv una partiala de AIS, care nu sunt asociate cu infertilitate 100% – probabil pentru ca testiculele continua sa isi pastreze o parte din functii.
Pacienta in sine a facut tratament amplu cu hormoni feminini pentru a isi dezvolta un uter foarte slab dezvoltat. Cum nu se mentioneaza nimic de o operatie de reconectare a uterului la vagin (majoritatea cazurilor de AIS complet au vaginul incomplet dezvoltat), presupun ca a avut noroc.
Acum, un pic de detalii:
Gena SRY (factorul de determinare testicular) este o gena prezenta pe cromozomul Y care codifica niste factori de transcriptie ce pornesc o cascada ce duce la diferentierea celulelor nediferentiate ale gonadelor fatului in testicule. Celulele Leydig si Sertoli se multiplica si isi incep activitatea secretorie. Celulele Leydig incep sa secrete testosteronul, cele Sertoli secreta hormonul antiMullerian – care opreste dezvoltarea canalelor lui Muller in trompele uterine, uter, col uterin si treimea superioara a vaginului.
Hormonii steroizi sexuali masculini sunt numiti si hormoni androgeni. Sunt responsabili pentru diferentierea si cresterea organelor sexuale masculine incepand cu saptamanile 6-7 de gestatie (cand celulele Leydig se activeaza). Printr-un proces mai complicat, complexul format din hormonii androgeni impreuna cu receptorii androgeni activeaza niste gene-tinta, care au un rol in dezvoltarea caracterelor sexuale masculine.
Diferentierea organelor genitale exterioare masculine (penis, scrot, uretra penisului) apare 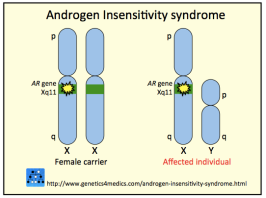 candva intre saptamanile 9 si 13 de gestatie si se intampla doar in conditiile prezentei unei concentratii adecvate de testosteron, urmata de conversia acestuia intr-un hormon mai potent, dihidrotestosteron, prin actiunea unei enzime in celulele testurilor-tinta. Pentru a isi face efectul, asa cum am zis, testosteronul si dihidrotestosteronul au nevoie de prezenta unor receptori pentru androgeni (AR) functionali. Orice disfunctie (cauzata sau nu de o mutatie in gena care codifica AR) in perioada aceasta (s. 9-13) in functionarea receptorilor pentru androgeni va duce la subexprimarea sau inexprimarea totala a caracterelor masculine si la dezvoltarea caracterelor feminine.
candva intre saptamanile 9 si 13 de gestatie si se intampla doar in conditiile prezentei unei concentratii adecvate de testosteron, urmata de conversia acestuia intr-un hormon mai potent, dihidrotestosteron, prin actiunea unei enzime in celulele testurilor-tinta. Pentru a isi face efectul, asa cum am zis, testosteronul si dihidrotestosteronul au nevoie de prezenta unor receptori pentru androgeni (AR) functionali. Orice disfunctie (cauzata sau nu de o mutatie in gena care codifica AR) in perioada aceasta (s. 9-13) in functionarea receptorilor pentru androgeni va duce la subexprimarea sau inexprimarea totala a caracterelor masculine si la dezvoltarea caracterelor feminine.
Printre problemele demne de mentionat ce urmeaza ar fi dezvoltarea partiala a testiculelor, urmata de necoborarea lor (ceea ce se numeste criptorhidie – am scris si eu despre asta). Unii pacienti sunt diagnosticati cu AIS dupa o operatie de excizie a unei hernii inghinale in care se elimina fix niste testicule necoborate. Testiculele necoborate au un risc asociat de cancer testicular, deci cred ca culmea cruzimii sortii ar fi pentru acesti oameni faptul ca sunt barbati (genetic), arata ca o femeie dar pot face cancer la testicule.
O alta observatie interesanta ar fi faptul ca testosteronul secretat (la pacientii ale caror testicule necoborate inca isi pastreaza o parte din functie) se poate transforma in estrogen in testuri, in anumite conditii, sub actiunea unei enzime, in procesul de aromatizare – de aici si dezvoltarea unor anumite caracteristici feminine.
Ei bine, copiii cu AIS complet cresc ca niste fete normale pana la pubertate, cand nu vor avea menarha (prima mensa). Diagnosticul se face tarziu sau foarte tarziu (cum a fost cazul pacientei din articol), pe baza caracteristicilor clinice (amenoree, lipsa parului pubian sau axilar, vagin scurtat, lipsa colului uterin si a uterului dar dezvoltarea normala a sanilor), investigatiilor de laborator (cariotipare, sau masurarea unor hormoni in sange: HCG, DHT, testosteron etc.) sau imagistice (IRM sau CT).
O ultima observatie ar fi ca diagnosticul de AIS complet a fost (din ce am citit eu) multi ani ascuns pacientilor descoperiti tarziu. Se presupunea ca socul ar fi fost prea mare, oferindu-li-se alte explicatii medicale pentru sterilitate (sau chiar pentru operatiile de extirpare de testicule).
Si, la final, dupa ce am trecut prin detaliile astea, explicatia pentru cum a nascut un barbat 2 gemeni: la un scan i s-a descoperit un uter foarte putin dezvoltat. Asa cum am zis, a urmat un tratament intensiv hormonal pentru dezvoltarea uterului, urmat de o procedura de fertilizare in vitro a 2 ovule recoltate de la un donator (2 viabile din 13 donate in total) in Cipru, care a culminat cu o nastere inainte de termen – indusa de doctori probabil din cauza dimensiunii uterului. Asa cum se intampla intr-un procent insemnat de fecundari in-vitro, ambele ovule s-au fecundat, de aici si norocul.
Cred ca e important de mentionat faptul ca femeia cu pricina a avut marele noroc de-a-si gasi un partener de viata care sa ignore problema genetica, cu care s-a si casatorit si care (din cate inteleg) va fi si tatal celor 2 gemeni.
Date luate din 2 articole (care au informatii suplimentare + cate un caz) de pe PubMed: 1 si 2.
Imagini cu cuplul fericit si copii in linkurile pentru articole, cateva imagini mai specializate in cele 2 articole PubMed.
O mentiune speciala pentru cine e cu adevarat curios: in afara de femei care sunt genetic (XY) barbati (AIS) mai avem si barbati care sunt genetic femei (XX), dar au prezenta gena SRY care determina sexul prezenta pe un cromozom X. La aberatii genetice care afecteaza cromozomii X si Y mai avem si sindromul Klinefelter (XXY), XYY, XXYY, trisomie X (XXX), XXXX si XXXXX. Cu exceptia celor cu XYY, toti sunt sterili (si ultimele tipuri de mutatii sunt extrem de rare).
P.S. In caz ca nu e clar, asa imi completez si recapitulez eu cunostinte de anatomie, boli, diagnostice si altele. Daca observa cineva ceva gresit, e liber, ba chiar il rog sa-mi atraga atentia. 🙂
P.P.S Ma intreb daca atleta sud-africana care a generat niste controverse acum cativa ani cu aspectul sau masculin suferea de o forma partiala de AIS – desi pare putin probabil ca insensibilitatea partiala la testosteron sa se manifeste atat de selectiv.